

Introduction
Treatment response of P. falciparum malaria is influenced by many factors. Such factors include drug quality, pharmacokinetic characteristics of individual drugs, parasite sensitivity, host genetics,1 drug–drug interactions and food–drug interactions. Inter-individual variability with respect to extent and rate of absorption, metabolism, distribution, plasma protein binding and elimination has been shown to influence the plasma concentration of drugs, hence affecting treatment outcomes in return.2 The inter-individual variability is common in Africa due to genetic diversity and heterogenicity.3 The complex patterns of population expansion, migration, contraction and admixture during evolutionary history explain the diversity observed in African populations.4 Sub-Saharan Africa (SSA) uniquely bears the highest global disease burden of infectious diseases, particularly malaria and HIV (90% and 69%, respectively). Unfortunately, only about 3% of patients with African or SSA genetic background take part in clinical trials globally.5 This implies that drugs employed in clinical practice lack information on safety and efficacy in African populations, thus the information employed in SSA relies mainly on post-marketing surveillance. Employing information from clinical trials which do not involve African populations creates uncertainty regarding the impact of genetic diversity during treatment. Thus, characterization of different ethnic groups in SSA is pivotal for implementation of pharmacogenomics or implementation of treatment based on the major drug metabolizer genotypes in the region.
The role of heritable genetic variations on treatment outcomes in malaria has gained much attention over recent years. However, such studies began in the 1950s with a remarkable finding on higher occurrence of hemolytic anaemia among black American soldiers than their white counterparts when primaquine was administered.6
The pattern of genetic variants affecting efficacy and safety of commonly used drugs in clinical practice has been of major interest in pharmacogenetic studies in SSA. Though not all populations have been studied, there has been progress as various genome initiatives, such as malariaGEN, African pharmacogenomic consortium (APC), H3Africa and African Genome variation Project have been established.7 Globally, significant advances have been achieved whereby individualized therapy is recommended for anticoagulants (warfarin-CYP2C9*2&*3), antiplatelet agents (clopidogrel- CYP2C19*2), antipsychotics (amitriptyline-CYP2D6&citalopram-CYP2C19)8 and anticancer treatments (tacrolimus-CYP3A5).9 With regard to P. falciparum malaria, CYP450 genotype-based treatment has not been introduced. However, G6PD deficiency testing prior to primaquine treatment for P. vivax malaria patients is now common practice in some South East Asian countries.10
Genetic variability in cytochrome P450 (CYP450) enzyme family has been shown to determine pharmacokinetic profiles of many drugs, including the antimalarials.11 The CYP3A4 gene, which is located on chromosome 7q21.3-q22.1 consisting of 13 exons,12 contributes to metabolism of about 50% of drugs used in clinical practice.13–15 In the liver microsomes, CYP3A4 mediates its reactions through the nicotinamide adenine dinucleotide phosphate-dependent electron pathway.12 The major contributor in drug metabolism in the CYP3A4 family is CYP3A4*1B (rs2740574),16 which is a result of A to G transition at nucleotide 392 in the promoter sequence of the gene.17 This single nucleotide polymorphism (SNP) results in poor metabolism of artemether and lumefantrine.11,18
CYP3A5 isoenzyme is regarded as the second contributor to drug metabolism after CYP3A4.19,20 CYP3A5*3 (rs776746) SNP occurs at highest abundance and plays a major role in drug metabolism within the CYP3A5 gene. This SNP is a result of the replacement of a nucleotide A by nucleotide G at locus 6986 within intron 3, producing an mRNA splice defect and consequently producing a premature stop codon.19,20 The CYP3A5*3 plays a significant role in the metabolism of artemether, lumefantrine, mefloquine, primaquine and chloroquine.4
The expression of both CYP3A4 and CYP3A5 is inducible by drugs. The increased enzyme activity is a result of increased expression via nuclear receptors pregnane X receptor (PXR), glucocorticoid receptor (GR), constitutive androstane receptor-β (CAR), vitamin D receptor (VDR) and hepatocyte nuclear factor-4 (HNF4α).21,22 These nuclear receptors increase transcription and expression of CYP3A4/5 after binding to DNA segments present in the CYP3A promoter (for CYP3A4) region, mainly PXR responsive element (prPXRE), xenobiotics responsive enhancer module (XREM) and constitutive liver enhancer module (CLEM4).
CYP2C8*2 SNP exists at higher frequencies among African than Asian and Caucasian populations. Unlike CYP2C8*2, the CYP2C8*3 SNP is common in Caucasians and Asians, but very rare in Africans.23 Both CYP2C8*2 and CYP2C8*3 are associated with a significant reduction in amodiaquine (AQ) in vitro metabolism. AQ adverse events are rare (1:2000) but very serious. These adverse drug reactions (ADRs) include neutropenia effects and severe liver failure. Studies associate these ADRs with a highly reactive and short-lived quinine-imine (QI) species which are products of AQ and DEAQ (metabolite of AQ) metabolism.24 CYP1A1 and CYP1B1 enzymes have been shown to play a great role in the formation of QI from in vitro biotransformation of AQ and DEAQ,25 since CYP1A1 and CYP1B1 are extra-hepatic localized, this could explain the occurrence of neutropenia effects observed with amodiaquine since the biotransformation of AQ and DEAQ to QI occurs in blood and not in the liver.24 There is a high possibility that CYP1A1 and CYP1B1 fast metabolizers are likely to suffer from AQ therapy side effects.24
CYP2A6 and CYP2B6 play a minor role in the biotransformation of artemisinin derivatives to form dihydroartemisinin, which is an active metabolite.26 The low activity of CYP2A6*1B and CYP2B6*6 is suggested to predict low plasma concentrations of dihydroartemisinin.26,27 The elimination of dihydroartemisinin depends on its conversion of inactive glucuronide conjugates, which is mediated by the highly polymorphic uridine diphosphate glucuronosyltransferase (UGT1A6 &UGT2B7) enzymes.28 A summary of the human genetic variants important for antimalarial drug metabolism is shown in Table 1.
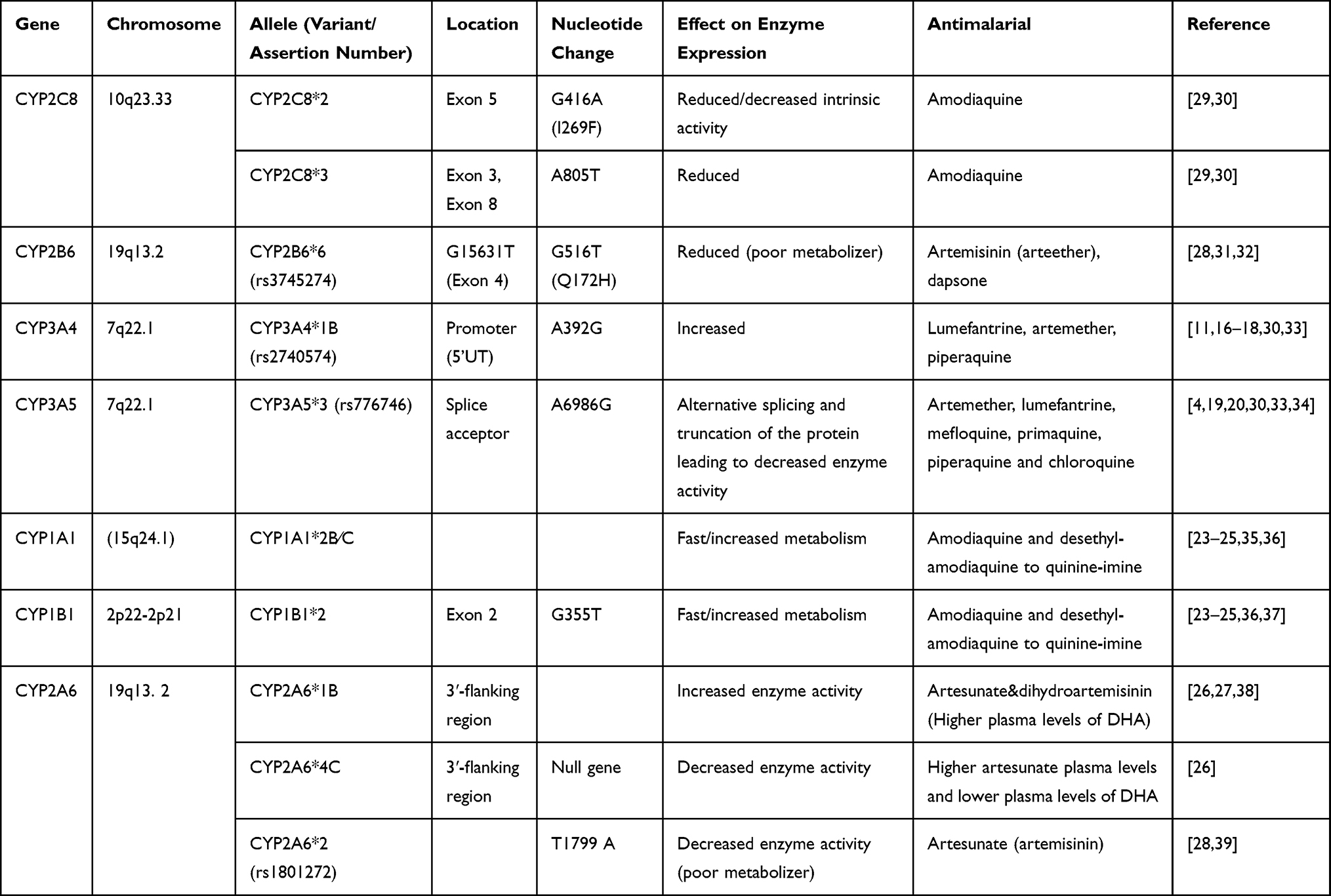 |
Table 1 CYP450 Single Nucleotide Polymorphisms Involved in Antimalarial Drugs Metabolism |
We have previously reported the frequencies of cytochrome P450 polymorphisms responsible for metabolism of antimalarial drugs in Africa.40 Other researchers have also extensively assessed these frequencies in the region.22,41,42 In general, the most frequently recorded SNPs are CYP2C8*2 (15–22%), CYP2B6*6 (30–50%), CYP3A4*1B (50–80%) and CYP3A5*3 (15–80%). However, the information on the influence of these polymorphisms on metabolism of antimalarial drugs used in clinical settings in SSA is scanty, thus evidence on the impact of these SNPs on pharmacokinetic profiles, efficacy and safety is not established. In this review, we explore and synthesize available evidence on the influence of cytochrome P450 polymorphisms on pharmacokinetic profiles and treatment outcomes of ACTs and other antimalarial drugs employed in SSA.
Methods
Literature Search
Literature search for published studies assessing the influence of CYP450 enzymes on PK profiles (plasma concentrations), efficacy and safety of antimalarial drugs in SSA was done through the Cochrane Central Register of Controlled Trials (CENTRAL), Google Scholar, EMBASE, SCOPUS, PubMed, Medline and LILACS online databases.
The search terms used were: ((“amodiaquine” and “CYP2C8*2”) AND (“efficacy”)) OR ((“amodiaquine” and “CYP2C8*2”) AND (“efficacy”) AND (“Africa”)) OR ((“amodiaquine” and “CYP2C8*2”) AND (“safety”) AND (“Africa”)) OR ((“amodiaquine” and “CYP2C8*2”) AND (“Pharmacokinetics”) AND (“Africa”)) OR ((“amodiaquine” and “CYP2C8*2”) AND (“Plasma concentration”) AND (“Africa”)). For lumefantrine, the search terms used were “CYP3A4*1B”) AND (“efficacy”)) OR ((“lumefantrine” and “CYP3A4*1B”) AND (“efficacy”) AND (“Africa”)) OR ((“lumefantrine” and “CYP3A4*1B”) AND (“safety”) AND (“Africa”)) OR ((“lumefantrine” and “CYP3A4*1B”) AND (“Pharmacokinetics”) AND (“Africa”)) OR ((“lumefantrine” and “CYP3A4*1B”) AND (“Plasma concentration”) AND (“Africa”)). The same approach was used for other drugs by replacing the name of the drug and the CYP450 SNP involved in metabolism of the drug. We used the Preferred Reporting Items for Systematic review and Meta-Analysis Protocols (PRISMA-P) 2015 checklist43 to identify studies to be included in our review.
Inclusion Criteria
Publications assessing the impact of CYP450 polymorphisms on PK profiles, safety and efficacy of antimalarials used for treatment of uncomplicated P. falciparum malaria in SSA were included. These studies were not time bound.
Exclusion Criteria
We excluded, for numerous reasons, studies assessing the influence of CYP450 SNPs on other drugs apart from antimalarials, studies assessing the influence of CYP450 on interaction between antiretroviral drugs (ART) and antimalarials excepting those with data for controls who are not on ART, studies assessing the impact of Phase II metabolizing enzymes on safety, efficacy and pharmacokinetic profiles, studies assessing the influence of drug transporters on pharmacokinetic profiles and treatment outcomes and studies assessing the influence of CYP450 polymorphisms on antimalarial treatment outcomes and PK profiles in regions other than SSA. Meeting communications and findings published based on animal models were not regarded as sufficient evidence and thus were also excluded. Studies assessing the influence of CYP450s on primaquine among P. vivax patients and volunteers were also excluded.
Methodological and Data Quality Assessment
Methodological quality assessment was done using the national institute of health (NIH) study quality assessment tools for controlled intervention, observational cohort and cross-sectional studies.44 The score ranged from 0 to 14, with a score of one point each that was then converted to percentages. The score range of 0–60% was regarded as low quality, 61–80% good quality and 81–100% excellent quality. Any difference in opinion with regard to extracted data and methodological quality assessment was resolved by consensus between the two independent reviewers. All included studies were of good to excellent quality as per the NIH scale.
Data Extraction
Two independent reviewers participated in the data extraction and screening of the results of the literature search and selected studies as per the inclusion criteria. Differences in opinion between reviewers on inclusion of studies were resolved through discussion. The basic information extracted included the author names, country in which the study was carried out, study population, sample size, SNPs, study endpoints, influence on pharmacokinetic profiles, influence on efficacy and influence on safety. Data was entered into extraction sheets.
Results
Study Characteristics
Fifty-six articles were included for full-text review from the 298 records (after removal of duplications) which were identified through the electronic database. Thirteen studies were finally included in data extraction after meeting the inclusion criteria. Details of the study search are shown in Figure 1. These studies originated from eight different countries within SSA.
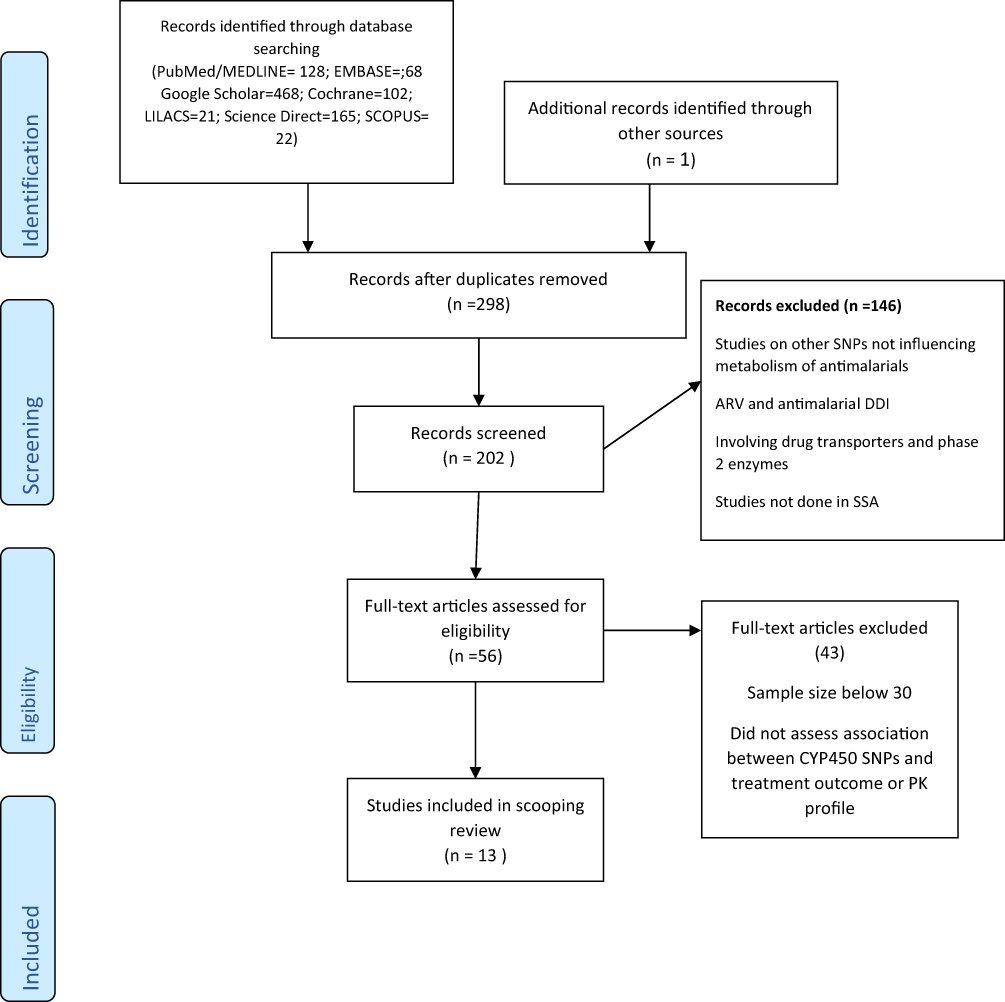 |
Figure 1 PRISMA flow diagram for article searches and screening. Note: Adapted from Moher D, Liberati A, Tetzlaff J, Altman DG, PRISMA Group* T. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. Annals of internal medicine.2009 Aug 18;151(4):264-9.43 |
Treatment Outcome
Pernaute-Lau et al45 carried out a study to assess the influence of CYP2C8*2 (805A>T) and CYP2C8*3 (416A>G) on treatment outcomes and tolerability among P. falciparum malaria patients treated with artesunate amodiaquine (ASAQ) in a Zanzibar population, Tanzania. This study reported presence of CYP2C8*3, which is rare in African populations but common among whites and Caucasians. The study end points were adverse events, ACPR, recrudescence and re-infection among malaria patients after 28- and 42-day follow up. There was no significant difference in recrudescence between subjects carrying CYP2C8*2 mutant alleles and wild type alleles. Carrying CYP2C8*2 or CYP2C8*3 did not predict for ACPR and re-infection among malaria patients. However, carrying CYP2C8*2 or CYP2C8*3 was associated with occurrence of non-serious adverse events compared to those with wild type alleles (Table 2).
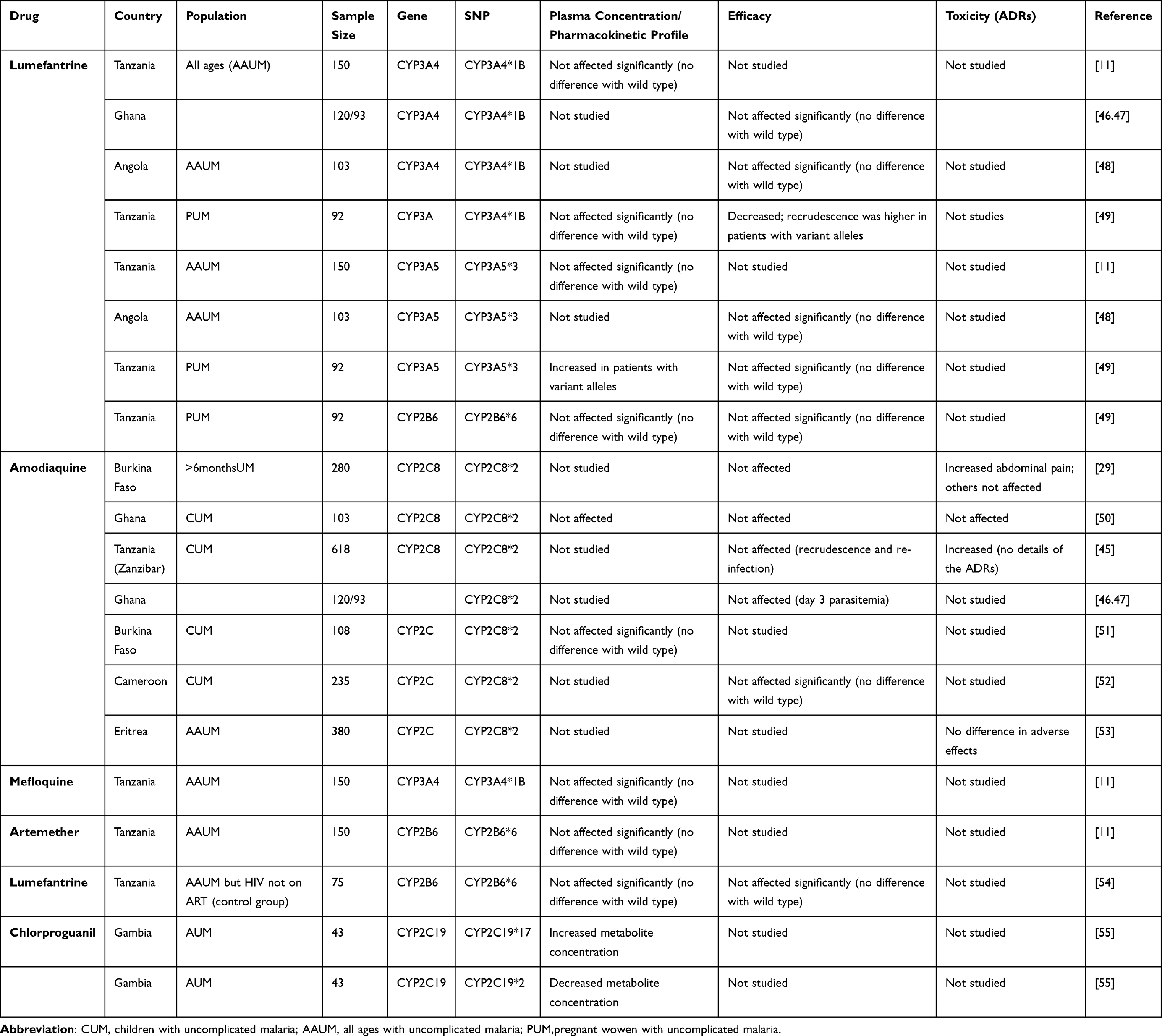 |
Table 2 Influence of CYP450 SNPs on Plasma Concentration and Treatment Outcomes |
Habtemikael et al53 carried out a cross-sectional study among malaria patients on artesunate amodiaquine in Eritrea. This study reported lack of association between CYP2C8*2 (805A>T) or CYP2C8*3 (416A>G) and extra pyramidal effects among Eritreans.
Mutagonda et al49 did a prospective cohort study in Tanzanian pregnant women to assess the influence of pharmacogenetics on day 7 plasma concentration and treatment outcomes. This study did not report association between CYP2B6*6 (516G>T), CYP3A5*3 (6986A>G) and day 28 ACPR. Unexpectedly, the study reported association between CYP3A4*1B (392A>G) with day 28 ACPR but not day 7 lumefantrine plasma concentration. This finding may have happened by chance since it is the day 7 plasma concentration of the drug which is suggested to determine treatment outcomes in malaria patients (Table 2).
Adjei et al50 assessed the influence of CYP450 polymorphisms on treatment outcomes and adverse events among Ghanaian children with uncomplicated malaria receiving ASAQ or AQ. No difference in efficacy and adverse events was observed between patients with CYP2C8*2 mutant allele and those with wild type allele (Table 2).
The assessment on the impact of CYP2C19*2 among Gambian adults with uncomplicated receiving chlorproguanil by Janha et al55 reported lack of significance in area under the curve (AUC) and maximum plasma concentration (Cmax) between those with loss of function allele and those without. Full pharmacokinetic sampling was done after three daily doses and plasma concentrations were used to determine AUC and Cmax (Table 2).
Another study in Burkina Faso, by Parikh et al,29 assessed the influence of CYP2C8 genotypes on efficacy and adverse events among malaria patients with uncomplicated malaria receiving ASAQ after 28 days of follow up. In this study, treatment outcomes in terms of ACPR, recrudescence and re-infection did not vary between patients with CYP2C8*2 (805A>T) and wild alleles (Table 2).
The influence of CYP2B6 genotypes on treatment outcomes among HIV-positive patients in the absence of antiretroviral therapy (ART) co-treatment (control group) was studied in Tanzania by Maganda et al.54 This group included only HIV-infected patients on artemether lumefantrine but who had not started taking ART, thus the drug–drug interactions between lumefantrine and efavirenz which could affect the plasma concentrations of either drug were not observed. The major finding was lack of association between CYP2B6*6 genotypes with incidence of recurrent parasitaemia (Table 2).
Mballa et al52 evaluated the influence of CYP2C8*2 variant allele on treatment outcomes among children with uncomplicated P. falciparum malaria in Cameroon. This study also reported lack of influence on treatment outcomes associated with CYP2C8 genotypes.
Lumefantrine and Amodiaquine Plasma Concentrations
Some et al51 did a cross-sectional survey on the influence of CYP2C8*2 (805A>T) SNPs on amodiaquine metabolism in Burkina Faso. There was no difference in day 7 DEAQ plasma concentrations between those with homozygous wild type allele and those with mutant allele. CYP2C8 genotypes did not contribute to differences in DEAQ concentration between subjects with mutant alleles and those with wild type alleles in Ghanaian children receiving ASAQ and AQ50 (Table 2).
Mutagonda et al49 also reported lack of association between CYP2B6*6 (516G>T) and day 7 lumefantrine concentration among pregnant women with uncomplicated P. falciparum malaria. Similar findings are reported for CYP3A4*1B (392A>G). However, patients with CYP3A5*3 (6986A>G) genotype had slightly higher day 7 lumefantrine concentration than their counterparts (CYP3A5*1/1*) but treatment outcomes were not affected (Table 2).
Adjei et al50 assessed the influence of CYP450 polymorphisms on pharmacokinetics among Ghanaian children with uncomplicated malaria receiving ASAQ or AQ. CYP2C8 genotypes did not contribute to the difference in the mean day 3 DEAQ concentrations between subjects with mutant and wild type allele.
Another study in Burkina Faso, by Parikh et al,29 evaluated the influence of CYP2C8 genotypes on efficacy and pharmacokinetic profiles of ASAQ among malaria patients with uncomplicated malaria. In this study, the intrinsic clearance of AQ for CYP2C8*2(805A>T) was six-fold lower than that of wild type allele (Table 2).
A population pharmacokinetics study was done in Tanzania by Hodel et al.11 The study population was 150 Tanzanian patients with uncomplicated malaria treated with ALU. Cambodian patients assessed for ASAQ were not discussed in this review. Neither CYP3A4*1B nor CYP3A5*3 affected lumefantrine plasma concentrations significantly. Similar findings were observed for artemether, whereby patients with CYP3A4*1B and CYP3A5*3 did not have different plasma concentration values compared to their counterparts (patients with wild alleles).
The influence of CYP2B6, CYP3A4 and CYP3A5 genotypes on day 7 lumefantrine plasma concentrations among HIV-positive patients in the absence of ART co-treatment (control group) was also studied in Tanzania, by Maganda et al.54 This group included only HIV-infected patients on artemether lumefantrine who had not started taking ART, thus the drug–drug interactions between lumefantrine and efavirenz, which could affect the plasma concentrations of either drug, were not observed. CYP2B6*6, CYP3A4*1B and CYP3A5*3 genotypes did not influence day 7 lumefantrine concentrations significantly among this group of malaria patients (Table 2).
There are studies which report association between drug levels and treatment outcomes but did not assess CYP 450 polymorphisms. Since CYP450 polymorphisms affect treatment outcomes through influencing plasma concentrations, it is worth considering these studies (Table 3). The above studies report lack of correlation between day 7 lumefantrine concentration cut-off values and treatment outcomes in SSA.
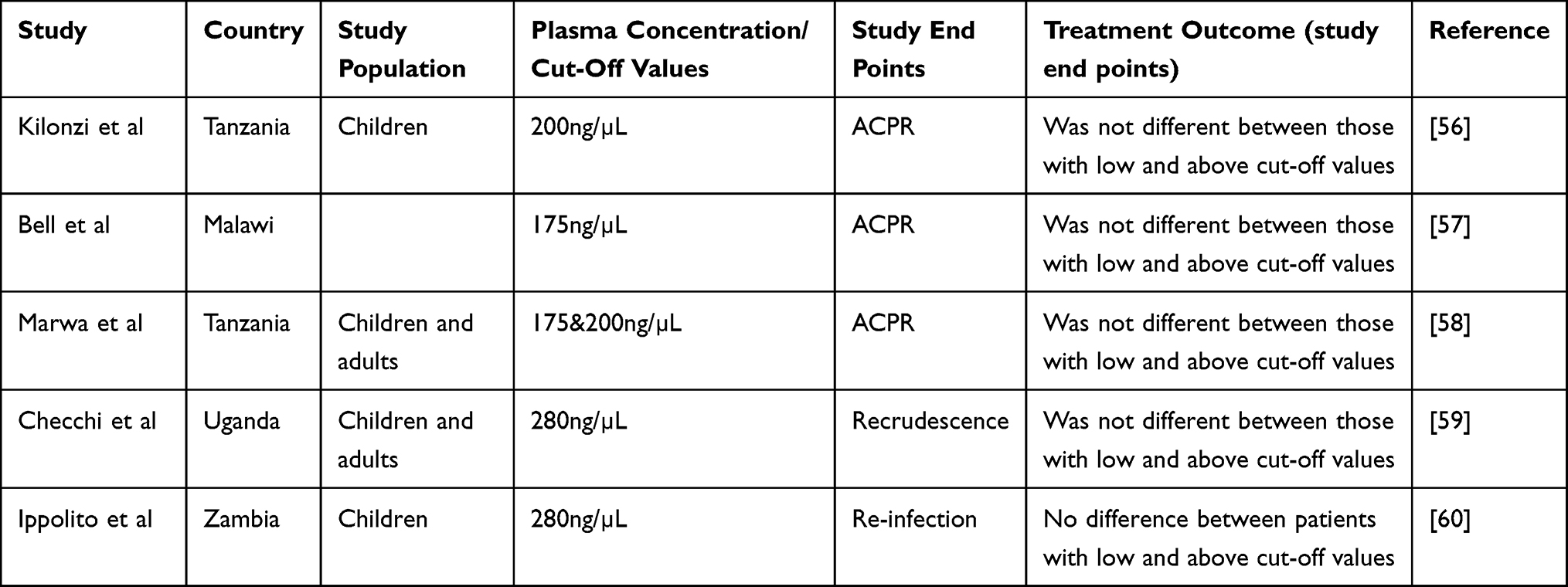 |
Table 3 Influence of Lumefantrine Plasma Concentration Below Cut-off Values on Treatment Outcome in SSA Populations |
Discussion
The major focus in studying CYP450 polymorphisms has been attainment of personalized medicine among patients. However, most studies only describe frequencies of CYP 450 variant alleles in different populations without assessing the effect of these polymorphisms on pharmacokinetics, efficacy and safety within populations. This review highlights evidence on the influence of CYP450 enzyme polymorphisms on antimalarial drug plasma concentrations and treatment outcomes among P. falciparum malaria patients in SSA.
The major CYP450 enzyme SNPs suggested to influence plasma concentrations and treatment outcomes (CYP3A4*1B, CYP3A5*5, CYP2B6*6 and CYP2C8*2) donot affect antimalarial drug plasma concentrations significantly in SSA, as shown in Table 2. In general, there was no difference in PK profiles between uncomplicated P. falciparum malaria patients with the mentioned CYP450 mutant alleles and those with wild type alleles in the region (Table 2). A similar finding was observed with antimalarial drug efficacy. These SNPs did not predict for low or high efficacy among patients with uncomplicated P. falciparum malaria. Findings from our review also suggest that a difference in ADRs between uncomplicated P. falciparum malaria patients with the CYP3A5*3, CYP3A4*1B and CYP2B6*6 SNPs and those with wild type alleles does not exist, with the exception of the CYP2C8*2 variant allele whereby a difference in minor adverse effects was observed in two studies. No difference was observed in terms of serious ADRs between subjects with CYP2C8*2 and those with wild type allele.
Lack of the influence of CYP450 polymorphisms on plasma concentrations, efficacy and serious adverse events suggests that dose optimization may not be necessary among P. falciparum malaria patients with CYP450 allelic variants in the region. Hodel et al also had a similar opinion to ours about ten years ago after employing genetic-based population pharmacokinetic modelling.11 Then, there were few studies on frequencies of genetic variants affecting the metabolism of antimalarial drugs in SSA. There has been advancement in terms of evidence on the presence of genetic variants in the region since Hodel et al suggested such findings. Our review provides a broader picture on the influence of CYP450 polymorphisms in treatment outcomes of P. falciparum malaria patients in various countries in SSA.
The suggestion that CYP450 genotyping-based treatment (tailored therapy) for uncomplicated P. falciparum malaria patients may not be of substantial worth is further supported by the recent findings in some SSA populations whereby lumefantrine plasma levels below cut-off values (<175ng/µL, 200ng/µL and 280ng/µL), which are suggested to predict treatment outcomes, did not affect treatment outcomes in these populations (Table 3). Drug plasma levels depend on enzyme metabolism as one of the key determinants, which in turn determines treatment response. Therefore, genotyping for variant alleles and their correlation with plasma levels may not be of great importance in these populations due to lack of an association between drug plasma levels and treatment outcomes. We observed similar findings in our recent study on association between day 3 and 7 lumefantrine plasma concentrations and treatment outcomes among P. falciparum malaria patients in Tanzania.58 Although studies included in this review have shown a lack of association between day 3 and 7 lumefantrine plasma concentrations and treatment outcomes, the influence of metabolites such as desbutyl-lumefantrine (usually not measured) on treatment outcomes in SSA needs to be assessed.
The lack of predictive effect of CYP450 polymorphisms and day 7 lumefantrine concentration on treatment outcomes in SSA may be attributed to high parasite sensitivity existing in the SSA region despite a growing threat of spread of resistant parasite strains from GMS regions. The acquired immunity among malaria patients in SSA populations, where most areas are malaria endemic thus individuals are exposed to multiple infections, may also account for the insignificant effect of CYP450 polymorphisms and plasma concentrations below cut-off values on treatment outcomes in the region. Although, the observation that lumefantrine plasma concentrations below cut-off values do not affect treatment outcomes in the region is encouraging, the impact of sub-optimal concentrations exposure to parasites should be worrying as far as selection of resistant parasites is concerned.
Our review focuses on SSA where most malaria patients are immune. Therefore, reviews of other regions of the world (where patients are non-immune) on the influence of CYP450 polymorphisms on antimalarial drug plasma concentrations, safety and efficacy among patients with uncomplicated P. falciparum malaria are warranted.
Like any other study, our review is not devoid of limitations. First, we understand there could be other unpublished data which we could not assess during our online search. Second, the lack of a sufficient number of studies (only a few countries are represented) assessing the influence of CYP450 polymorphisms on pharmacokinetic parameters, efficacy and safety in SSA limits the power of our review and did not allow us to carry out a meta-analysis.
There could be other factors, such as polymorphism in Phase II SNP genes encoding N-Acetyl Transferase 2, drug transporters (such as ABCB1 [MDR1], ABCC2 [MRP2]) and drug targets, which may also influence antimalarial drug plasma concentrations, safety and cure rates among malaria patients, thus limiting the findings reported in this study. However, since we only considered CYP450s, these factors are regarded as constant and thus their influence may not vary. We understand polypharmacy is common in SSA, thus drug–drug interactions are common and could influence CYP450 expression and treatment outcomes among malaria patients. To minimize this, only studies which followed the WHO protocol for assessment of the efficacy of antimalarials were included and studies assessing drug–drug interactions among malaria patients were excluded. Despite these limitations, this review is unique because it is the first review to assess the influence of CYP450 polymorphisms on antimalarial drug plasma concentrations, safety and efficacy among P. falciparum malaria patients in SSA.
Conclusion
This review reports lack of influence of CYP3A5*3, CYP3A4*1B, CYP2C8*3 and CYP2B6*6 SNPs on plasma concentrations, efficacy and safety among P. falciparum malaria patients in the region. This suggests that CYP450 genotyping-based dose optimization (personalized medicine) may not be important in malaria patients with the variant alleles in SSA.
Abbreviations
ART, antiretroviral therapy; CYP450, cytochrome P450 polymorphisms; DDIs, drug–drug interactions; GMS, Great Mekong Sub-region; SSA, sub-Saharan Africa; PK, pharmacokinetics; HIV, Human Immunodeficiency Virus.
Funding
No grant was received. However, authors have been receiving salaries by their respective universities during the literature search and manuscript writing.
Disclosure
The authors declare that they have no competing interests in this work.
References
1. Obua C, Hellgren U, Ntale M, et al. Population pharmacokinetics of chloroquine and sulfadoxine and treatment response in children with malaria: suggestions for an improved dose regimen. Br J Clin Pharmacol. 2008;65(4):493–501. doi:10.1111/j.1365-2125.2007.03050.x
2. Pang KS. Modeling of intestinal drug absorption: roles of transporters and metabolic enzymes (for the Gillette Review Series). Drug Metabol Disposit. 2003;31(12):1507–1519. doi:10.1124/dmd.31.12.1507
3. Campbell MC, Tishkoff SA. African genetic diversity: implications for human demographic history, modern human origins, and complex disease mapping. Annu Rev Genomics Hum Genet. 2008;9:403–433. doi:10.1146/annurev.genom.9.081307.164258
4. Dandara C, Swart M, Mpeta B, Wonkam A, Masimirembwa C. Cytochrome P450 pharmacogenetics in African populations: implications for public health. Expert Opin Drug Metab Toxicol. 2014;10(6):769–785. doi:10.1517/17425255.2014.894020
5. Mpye K, Matimba A, Dzobo K, Chirikure S, Wonkam A, Dandara C. Disease burden and the role of pharmacogenomics in African populations. Global Health Epidemiol Genom. 2017;2. doi:10.1017/gheg.2016.21
6. Motulsky AG. Drug reactions, enzymes, and biochemical genetics. J Am Med Assoc. 1957;165(7):835–837. doi:10.1001/jama.1957.72980250010016
7. Dandara C, Masimirembwa C, Haffani YZ, et al. African Pharmacogenomics Consortium: consolidating pharmacogenomics knowledge, capacity development and translation in Africa: consolidating pharmacogenomics knowledge, capacity development and translation in Africa. AAS Open Res. 2019;2:19. doi:10.12688/aasopenres.12965.1
8. Dandara C, Matimba A. A Glimpse into Pharmacogenomics in Africa. Cambridge, UK: Cambridge University Press; 2019.
9. Tata BE, Ambele M, Pepper M. Barriers to implementing clinical pharmacogenetics testing in Sub-Saharan Africa. A critical review. Pharmaceutics. 2020;12(9):809. doi:10.3390/pharmaceutics12090809
10. Ley B, Luter N, Espino FE, et al. The challenges of introducing routine G6PD testing into radical cure: a workshop report. BioMed Central. 2015. doi:10.1186/s12936-015-0896-8
11. Staehli Hodel EM, Csajka C, Ariey F, et al. Effect of single nucleotide polymorphisms in cytochrome P450 isoenzyme and N-acetyltransferase 2 genes on the metabolism of artemisinin-based combination therapies in malaria patients from Cambodia and Tanzania. Antimicrob Agents Chemother. 2013;57(2):950–958. doi:10.1128/AAC.01700-12
12. Keshava C, McCanlies EC, Weston A. CYP3A4 polymorphisms—potential risk factors for breast and prostate cancer: a HuGE review. Am J Epidemiol. 2004;160(9):825–841. doi:10.1093/aje/kwh294
13. Seripa D, Pilotto A, Panza F, Matera MG, Pilotto A. Pharmacogenetics of cytochrome P450 (CYP) in the elderly. Ageing Res Rev. 2010;9(4):457–474. doi:10.1016/j.arr.2010.06.001
14. Thummel KE, Wilkinson GR. In vitro and in vivo drug interactions involving human CYP3A. Annu Rev Pharmacol Toxicol. 1998;38:389–430. doi:10.1146/annurev.pharmtox.38.1.389
15. Feltrin C, Farias IV, Sandjo LP, Reginatto FH, Simões C. Effects of standardized medicinal plant extracts on drug metabolism mediated by CYP3A4 and CYP2D6 enzymes. Chem Res Toxicol. 2020;33(9):2408–2419. doi:10.1021/acs.chemrestox.0c00182
16. Alessandrini M, Asfaha S, Dodgen TM, Warnich L, Pepper MS. Cytochrome P450 pharmacogenetics in African populations. Drug Metab Rev. 2013;45(2):253–275. doi:10.3109/03602532.2013.783062
17. El-Shair S, Al Shhab M, Zayed K, Alsmady M, Zihlif M. Association between CYP3A4 and CYP3A5 genotypes and cyclosporine’s blood levels and doses among Jordanian kidney transplanted patients. Curr Drug Metab. 2019;20(8):682–694. doi:10.2174/1389200220666190806141825
18. Piedade R, Gil JP. The pharmacogenetics of antimalaria artemisinin combination therapy. Expert Opin Drug Metab Toxicol. 2011;7(10):1185–1200. doi:10.1517/17425255.2011.608660
19. Eng H-S, Mohamed Z, Calne R, et al. The influence of CYP3A gene polymorphisms on cyclosporine dose requirement in renal allograft recipients. Kidney Int. 2006;69(10):1858–1864. doi:10.1038/sj.ki.5000325
20. Tang H-L, Ma -L-L, Xie H-G, Zhang T, Hu Y-F. Effects of the CYP3A5* 3 variant on cyclosporine exposure and acute rejection rate in renal transplant patients: a meta-analysis. Pharmacogenet Genomics. 2010;20(9):525–531. doi:10.1097/FPC.0b013e32833ccd56
21. Daly AK. Significance of the minor cytochrome P450 3A isoforms. Clin Pharmacokinet. 2006;45(1):13–31. doi:10.2165/00003088-200645010-00002
22. Saiz-Rodríguez M, Almenara S, Navares-Gómez M, et al. Effect of the most relevant CYP3A4 and CYP3A5 polymorphisms on the pharmacokinetic parameters of 10 CYP3A substrates. Biomedicines. 2020;8(4):94. doi:10.3390/biomedicines8040094
23. Daily EB, Aquilante CL. Cytochrome P450 2C8 pharmacogenetics: a review of clinical studies. Pharmacogenomics. 2009;10(9):1489–1510. doi:10.2217/pgs.09.82
24. Cavaco I, Piedade R, Msellem MI, Bjorkman A, Gil JP. Cytochrome 1A1 and 1B1 gene diversity in the Zanzibar islands. Trop Med Int Health. 2012;17(7):854–857.
25. Johansson T, Jurva U, Gronberg G, Weidolf L, Masimirembwa C. Novel metabolites of amodiaquine formed by CYP1A1 and CYP1B1: structure elucidation using electrochemistry, mass spectrometry, and NMR. Drug Metab Dispos. 2009;37(3):571–579. doi:10.1124/dmd.108.025171
26. Phompradit P, Muhamad P, Cheoymang A, Na-Bangchang K. Preliminary investigation of the contribution of CYP2A6, CYP2B6, and UGT1A9 polymorphisms on artesunate-mefloquine treatment response in Burmese patients with Plasmodium falciparum malaria. Am J Trop Med Hyg. 2014;91(2):361. doi:10.4269/ajtmh.13-0531
27. Yusof W, Hua GS. Gene, ethnic and gender influences predisposition of adverse drug reactions to artesunate among Malaysians. Toxicol Mech Methods. 2012;22(3):184–192. doi:10.3109/15376516.2011.623331
28. Kerb R, Fux R, Mörike K, et al. Pharmacogenetics of antimalarial drugs: effect on metabolism and transport. Lancet Infect Dis. 2009;9(12):760–774. doi:10.1016/S1473-3099(09)70320-2
29. Parikh S, Ouedraogo JB, Goldstein J, Rosenthal P, Kroetz D. Amodiaquine metabolism is impaired by common polymorphisms in CYP2C8: implications for malaria treatment in Africa. Clin Pharmacol Ther. 2007;82(2):197–203. doi:10.1038/sj.clpt.6100122
30. Tornio A, Backman JT. Cytochrome P450 in pharmacogenetics: an update. Adv Pharmacol. 2018;83:3–32.
31. Hoffman SMG, Nelson DR, Keeney DS. Organization, structure and evolution of the CYP2 gene cluster on human chromosome 19. Pharmacogenet Genomics. 2001;11(8):687–698. doi:10.1097/00008571-200111000-00007
32. Zanger UM, Klein K, Saussele T, Blievernicht J, Hofmann M, Schwab M. Polymorphic CYP2B6: molecular mechanisms and emerging clinical significance. Pharmacogenomics. 2007;8(7):743–759. doi:10.2217/14622416.8.7.743
33. Liu H, Xie Y, Cai T, Xing J. No effect of PXR (8055C> T) polymorphism on the pharmacokinetic profiles of piperaquine in healthy Chinese subjects. Curr Drug Metab. 2021;23(2):164–170.
34. Gim J-A, Kwon Y, Lee HA, et al. A machine learning-based identification of genes affecting the pharmacokinetics of tacrolimus using the DMETTM plus platform. Int J Mol Sci. 2020;21(7):2517. doi:10.3390/ijms21072517
35. Cavaco IdCL. Molecular determinants of the response to malaria therapeutics; 2007.
36. Gil JP. The pharmacogenetics of the antimalarial amodiaquine: citeseer; 2012.
37. Jin J, Lin F, Liao S, Bao Q, Ni L, Wei Q-Y. Effects of SNPs (CYP1B1* 2 G355T, CYP1B1* 3 C4326G, and CYP2E1* 5 G-1293C), smoking, and drinking on susceptibility to laryngeal cancer among Han Chinese. PLoS One. 2014;9(10):e106580. doi:10.1371/journal.pone.0106580
38. Mwenifumbo JC, Lessov‐Schlaggar CN, Zhou Q, et al. Identification of novel CYP2A6* 1B variants: the CYP2A6* 1B allele is associated with faster in vivo nicotine metabolism. Clin Pharmacol Ther. 2008;83(1):115–121. doi:10.1038/sj.clpt.6100246
39. Ezzeldin N, El-Lebedy D, Darwish A, et al. Association of genetic polymorphisms CYP2A6* 2 rs1801272 and CYP2A6* 9 rs28399433 with tobacco-induced lung Cancer: case-control study in an Egyptian population. BMC Cancer. 2018;18(1):1–9. doi:10.1186/s12885-018-4342-5
40. Marwa KJ, Schmidt T, Sjögren M, Minzi OM, Kamugisha E, Swedberg G. Cytochrome P450 single nucleotide polymorphisms in an indigenous Tanzanian population: a concern about the metabolism of artemisinin-based combinations. Malar J. 2014;13(1):1–7. doi:10.1186/1475-2875-13-420
41. Ferreira PE, Veiga MI, Cavaco I, et al. Polymorphism of antimalaria drug metabolizing, nuclear receptor, and drug transport genes among malaria patients in Zanzibar, East Africa. Ther Drug Monit. 2008;30(1):10–15. doi:10.1097/FTD.0b013e31815e93c6
42. Roederer MW, McLeod H, Juliano JJ. Can pharmacogenomics improve malaria drug policy? Bull World Health Organ. 2011;89:838–845. doi:10.2471/BLT.11.087320
43. Moher D, Shamseer L, Clarke M, et al. Preferred reporting items for systematic review and meta-analysis protocols (PRISMA-P) 2015 statement. Syst Rev. 2015;4(1):1. doi:10.1186/2046-4053-4-1
44. Health NIo. National Heart Lung, and Blood Institute. Study Quality Assessment Tools. Bethesda, MD, USA: National Institutes of Health; 2018.
45. Pernaute-Lau L, Morris U, Msellem M, Mårtensson A, Björkman A, Gil JP. Influence of cytochrome P450 (CYP) 2C8 polymorphisms on the efficacy and tolerability of artesunate‐amodiaquine treatment of uncomplicated Plasmodium falciparum malaria in Zanzibar. Malar J. 2021;20(1):1–7. doi:10.1186/s12936-021-03620-6
46. Hodoameda P. Determination of Plasmodium Falciparum and Host Genetic Factors That Affect the Efficacy of the Artemisinin-Based Combination Partner Drugs Used in Ghana. University Of Ghana; 2019.
47. Hodoameda P, Duah-Quashie NO, Hagan CO, et al. Plasmodium falciparum genetic factors rather than host factors are likely to drive resistance to ACT in Ghana. Malar J. 2020;19(1):1–8. doi:10.1186/s12936-020-03320-7
48. Kiaco K, Rodrigues AS, Do Rosário V, Gil JP, Lopes D. The drug transporter ABCB1 c. 3435C> T SNP influences artemether–lumefantrine treatment outcome. Malar J. 2017;16(1):1–6. doi:10.1186/s12936-017-2006-6
49. Mutagonda RF, Kamuhabwa AA, Minzi O, et al. Effect of pharmacogenetics on plasma lumefantrine pharmacokinetics and malaria treatment outcome in pregnant women. Malar J. 2017;16(1):1–10. doi:10.1186/s12936-017-1914-9
50. Adjei GO, Kristensen K, Goka BQ, et al. Effect of concomitant artesunate administration and cytochrome P4502C8 polymorphisms on the pharmacokinetics of amodiaquine in Ghanaian children with uncomplicated malaria. Antimicrob Agents Chemother. 2008;52(12):4400–4406. doi:10.1128/AAC.00673-07
51. Somé FA, Bazié T, Ehrlich HY, et al. Investigating selected host and parasite factors potentially impacting upon seasonal malaria chemoprevention in Bama, Burkina Faso. Malar J. 2020;19(1):1–8. doi:10.1186/s12936-020-03311-8
52. Mballa R, Chedjou J, Ngwafor R. Single nucleotide polymorphisms in the cyp2C8 and nat2 genes and treatment outcomes in patients suffering from uncomplicated malaria in Garoua, Northern Region of Cameroon. Pharm Pharmacol Int J. 2019;7(4):147–153.
53. Habtemikael L, Russom M, Bahta I, et al. Prevalence of CYP2C8* 2 and* 3 among Eritreans and its potential impact on artesunate/amodiaquine treatment. Pharmgenomics Pers Med. 2020;13:571. doi:10.2147/PGPM.S276215
54. Maganda B, Minzi O, Ngaimisi E, Kamuhabwa A, Aklillu E. CYP2B6* 6 genotype and high efavirenz plasma concentration but not nevirapine are associated with low lumefantrine plasma exposure and poor treatment response in HIV-malaria-coinfected patients. Pharmacogenomics J. 2016;16(1):88–95. doi:10.1038/tpj.2015.37
55. Janha RE, Sisay-Joof F, Hamid-Adiamoh M, et al. Effects of genetic variation at the CYP2C19/CYP2C9 locus on pharmacokinetics of chlorcycloguanil in adult Gambians. Pharmacogenomics. 2009;10(9):1423–1431. doi:10.2217/pgs.09.72
56. Kilonzi M, Minzi O, Mutagonda R, et al. Usefulness of day 7 lumefantrine plasma concentration as a predictor of malaria treatment outcome in under-fives children treated with artemether-lumefantrine in Tanzania. Malar J. 2020;19(1):1–8. doi:10.1186/s12936-020-3150-y
57. Bell DJ, Wootton D, Mukaka M, et al. Measurement of adherence, drug concentrations and the effectiveness of artemether-lumefantrine, chlorproguanil-dapsone or sulphadoxine-pyrimethamine in the treatment of uncomplicated malaria in Malawi. Malar J. 2009;8(1):1–11. doi:10.1186/1475-2875-8-204
58. Marwa KJ, Liwa AC, Konje ET, Mwita S, Kamugisha E, Swedberg G. Lumefantrine plasma concentrations in uncontrolled conditions among patients treated with artemether-lumefantrine for uncomplicated plasmodium falciparum malaria in Mwanza, Tanzania. Int J Infect Dis. 2022;123:192–199. doi:10.1016/j.ijid.2022.08.020
59. Checchi F, Piola P, Fogg C, et al. Supervised versus unsupervised antimalarial treatment with six-dose artemether-lumefantrine: pharmacokinetic and dosage-related findings from a clinical trial in Uganda. Malar J. 2006;5(1):1–8. doi:10.1186/1475-2875-5-59
60. Ippolito MM, Pringle JC, Siame M, et al. Therapeutic Efficacy of artemether–lumefantrine for uncomplicated falciparum malaria in Northern Zambia. Am J Trop Med Hyg. 2020;103(6):2224. doi:10.4269/ajtmh.20-0852